2025-05-17-scBridge
post on 17 May 2025 about 2380words require 8min
CC BY 4.0 (除特别声明或转载文章外)
如果这篇博客帮助到你,可以请我喝一杯咖啡~
2025-05-17-scBridge embraces cell heterogeneity in single-cell RNA-seq and ATAC-seq data integration
相关资料
https://www.nature.com/articles/s41467-023-41795-5
scBridge embraces cell heterogeneity in single-cell RNA-seq and ATAC-seq data integration - PubMed
https://github.com/GreenleafLab/MPAL-Single-Cell-2019
Motivation:
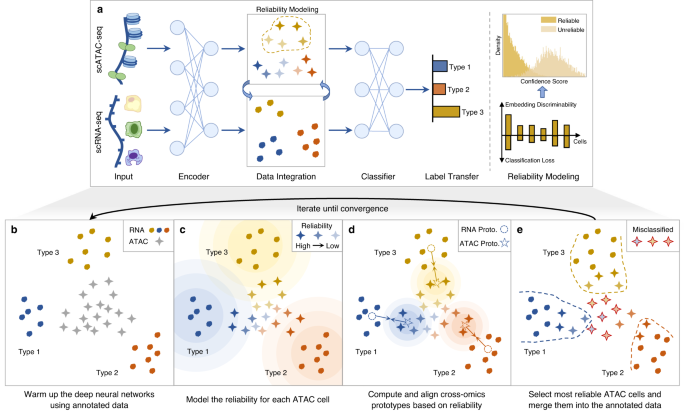
概述: scBridge 是一套面向单细胞多组学数据(scRNA-seq 与 scATAC-seq)整合的半监督异构迁移学习框架。它的核心策略是:先用标注的 scRNA-seq 数据“热身”(warm-up)一个深度编码器和分类器,然后根据每个 scATAC-seq 细胞与 RNA 原型的相似度和分类置信度,评估其在组学整合中的“可靠性”,再借助可靠的 ATAC 细胞对齐 RNA 原型;如此迭代,从“易”到“难”逐步缩小 RNA/ATAC 之间的调制差距。大量 benchmark 测试表明,scBridge 在嵌入质量、标签转移准确性和对注释稀缺、技术噪声的鲁棒性方面,均显著优于六种代表性方法(Harmony、Seurat、scJoint、Portal、GLUE、Conos)(Nature)。
单细胞多组学整合的必要性
- 单细胞 RNA 测序(scRNA-seq)可描绘基因表达全景,揭示细胞类型与状态的多样性;而单细胞 ATAC 测序(scATAC-seq)则刻画染色质可及性,用以探究基因调控机制(Frontiers)。
- 将两种组学 unpaired(非配对)数据整合,可从表达与调控层面构建更全面的细胞图谱,但它们在分布、稀疏性、噪声水平上差异巨大,给算法带来挑战(Nature)。
现有整合方法
- Harmony:通过批次校正,将不同数据集投影到同一低维空间,突出细胞类型聚类而非实验条件(PMC)。
- Seurat:基于互为最近邻的锚点(MNN)和标签转移(Transfer Anchors)实现多模态共嵌入(Satija Lab)。
- scJoint:半监督框架并行训练标注(scRNA)与未标注(scATAC)数据,完成标签转移和联合可视化(PMC)。
- GLUE:图模型理念,显式建模不同组学之间的调控网络,构建统一嵌入(Nature)。
- Conos:通过全局图结构将多样本 scRNA 数据联通,突出细胞类型一致性传播(GitHub)。
- Portal:基于对抗学习的半监督多组学整合管线(本文略)。
这些方法多对所有细胞“一视同仁”地整合,未利用组学内部的“异质性”信息来引导模型从“易”到“难”分层对齐。

Method:
异构迁移学习框架概览
scBridge 将标注的 scRNA-seq 与未标注的 scATAC-seq 数据看作源域与目标域,通过迭代的迁移学习流程逐步对齐两种组学的嵌入表示(Nature)。
- Warm-up(热身) 使用带标签的 scRNA-seq 数据训练一个深度编码器 $f$ 和分类器 $g$,并计算出初始的 RNA 嵌入及原型(每种细胞类型的平均嵌入)(Nature)。
- 可靠性建模 对每个 scATAC-seq 细胞,计算其嵌入与 RNA 原型的欧氏距离(可辨识度)及分类器损失(置信度),并用高斯混合模型估计每个细胞的“可靠性”(Nature)。
- 原型对齐 根据可靠性加权平均选出同类型 ATAC 细胞,构建 ATAC 原型,并将其与对应的 RNA 原型对齐(例如最小化原型间距离)(Nature)。
- 迭代融合 将本轮最“可靠”的 ATAC 细胞加入标注集中,附带当前预测标签,重复上述热身 → 建模 → 对齐过程,直至所有细胞被整合完毕(Nature)。
Result:
黄金 benchmark 验证
- 在三组“金标准”配对数据集(SNARE-seq 脑皮层、SHARE-seq 骨髓、10x Multiome 肾脏)上,虽然未使用配对信息,仅作验证,但 scBridge 在嵌入质量和标签转移上均领先六种基线方法(Nature)。
与主流方法对比
- 标签转移准确率:在 PBMC 数据集上,scBridge 平均比第二名 scJoint 提高约 5% 的准确率(p ≤ 1e−3)(Nature)。
- 鲁棒性测试:在稀缺注释(25%、50%、75%)和不同 dropout 强度下,scBridge 均表现出更稳健的 F1 分数和轮廓系数(Nature)。
- 可扩展性:由于仅需小批量优化,算法在细胞数量线性增长时占用常数内存,可轻松处理百万级别数据集(Nature)。
创新
- 细胞异质性利用:首次将组学内部的“易整合”细胞作为“桥梁”,显著提升跨模态对齐效果(Nature)。
- 从易到难的分层融合:通过迭代引入可靠细胞,降低了一次性对齐带来的误差累积,可有效应对噪声与注释不足问题(Nature)。
- 通用性强:框架输入只需保证行(细胞)一致,列(基因、蛋白等)需对齐,可推广到其他单细胞多组学场景(Nature)。
- 开源可复现:论文随文附带代码与数据,方便社区复现与扩展(Semantic Scholar)。
未来展望
- 更多模态融合:可直接应用于蛋白质组、表观组等其他单细胞组学,构建更全面的细胞图谱。
- 与自动注释工具结合:可将任何组学注释数据纳入 heterogenous learning 流程,进一步提升新型细胞类型发现能力。
- 下游分析拓展:整合后嵌入可无缝接入细胞谱系追踪、调控网络推断等多种后续分析管线,为生物学研究提供新范式。
Experiment:
我完成的任务与收获
- 重装服务器的 conda 环境
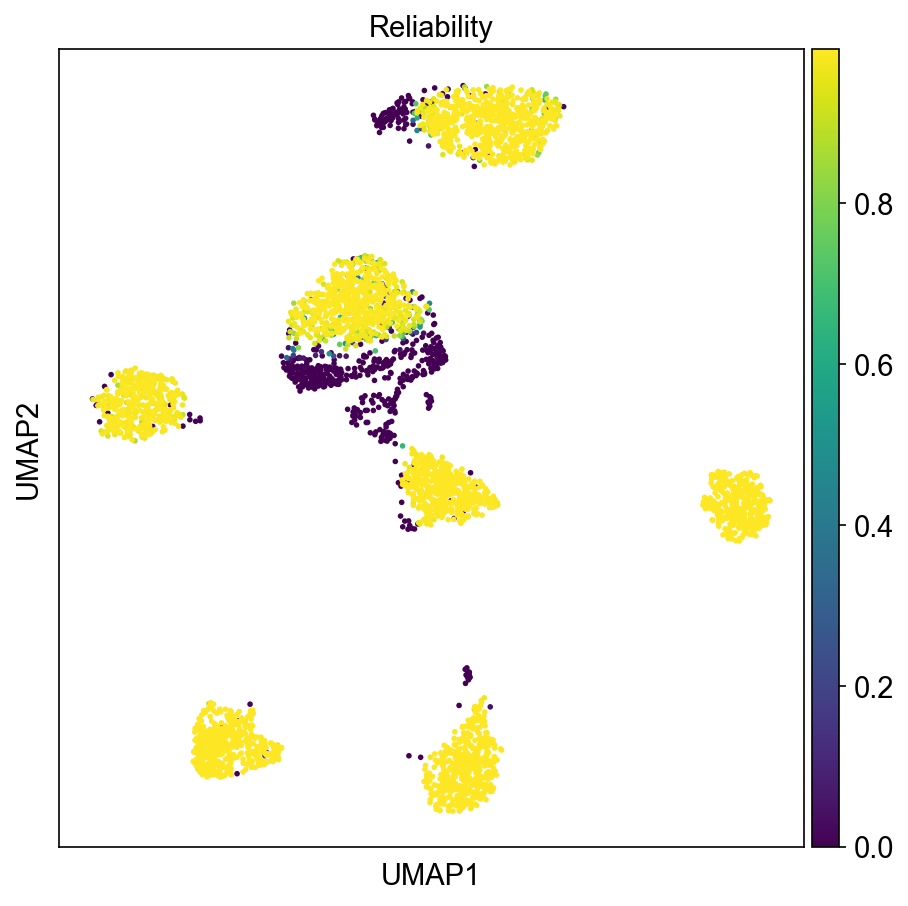
运行结果
umap_PBMC_CellType
umap_PBMC_Domain

umap_PBMC_Reliability

Related posts
Loading comments...